
The HECBioSim benchmark suite consists of a set of simple benchmarks for a number of popular Molecular Dynamics (MD) engines, each of which is set at a different atom count. Our benchmarking suite has now been automated by making use of our job submission tool Longbow, this allows us to quickly benchmark new code versions or to benchmark HPC machines that we have not previously done this for. Having such a benchmarking suite enables us to provide guidelines about run-times that members of our community can expect when applying for time on HPC facilities and allows us to assess such applications at a technical level when supporting an application on given hardware available to us.
The Benchmark Systems
The benchmark suite currently contains benchmarks for the AMBER, GROMACS, LAMMPS and NAMD molecular dynamics packages. The following systems have currently been setup for each of the MD packages to represent examples of the sorts of simulations that biosimulation scientists would do in production. Currently we only have benchmarks for classical MD, however, we are working on new benchmarks to widen our benchmark suite to include other techniques.

20K atom system - 3NIR Crambin
Total number of atoms = 19,605
Protein atoms = 642 Water atoms = 18,963
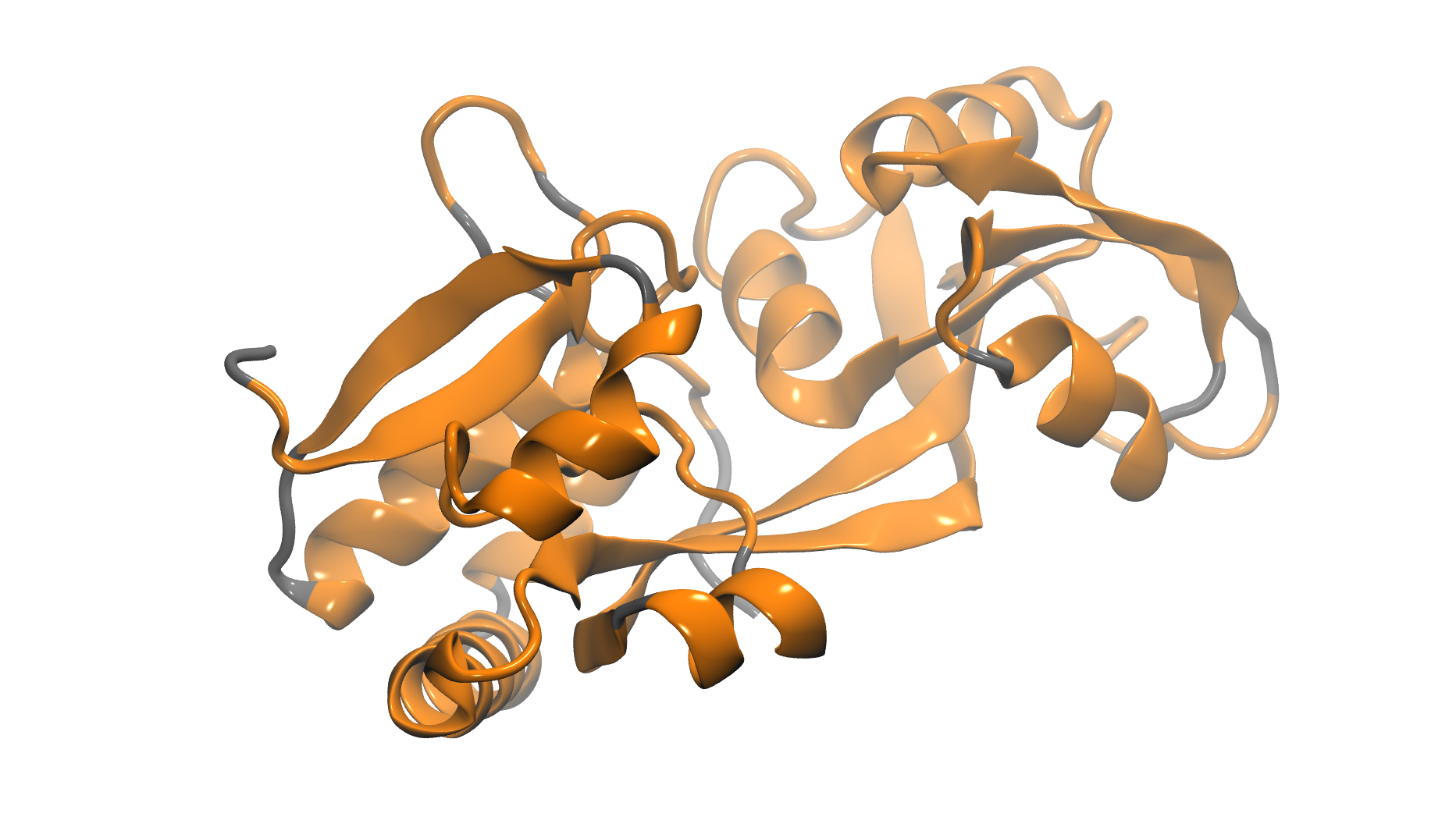
61K atom system - 1WDN Glutamine-Binding Protein
Total number of atoms = 61,153
Protein atoms = 3,555 Water atoms = 57,957 Ions = 1
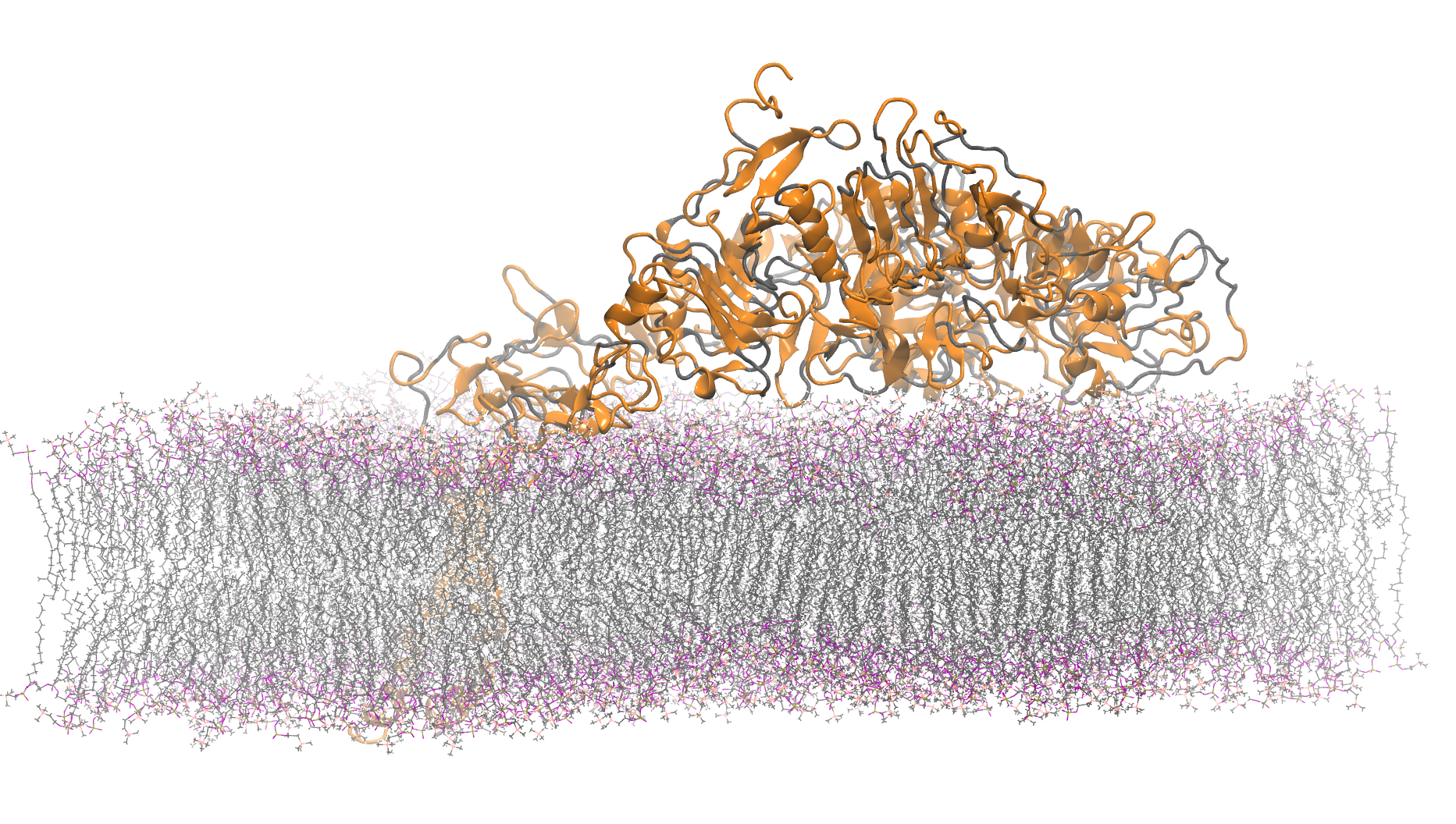
465K atom system - hEGFR Dimer of 1IVO and 1NQL
Total number of atoms = 465,399
Protein atoms = 21,749 Lipid atoms = 134,268 Water atoms = 309,087 Ions = 295
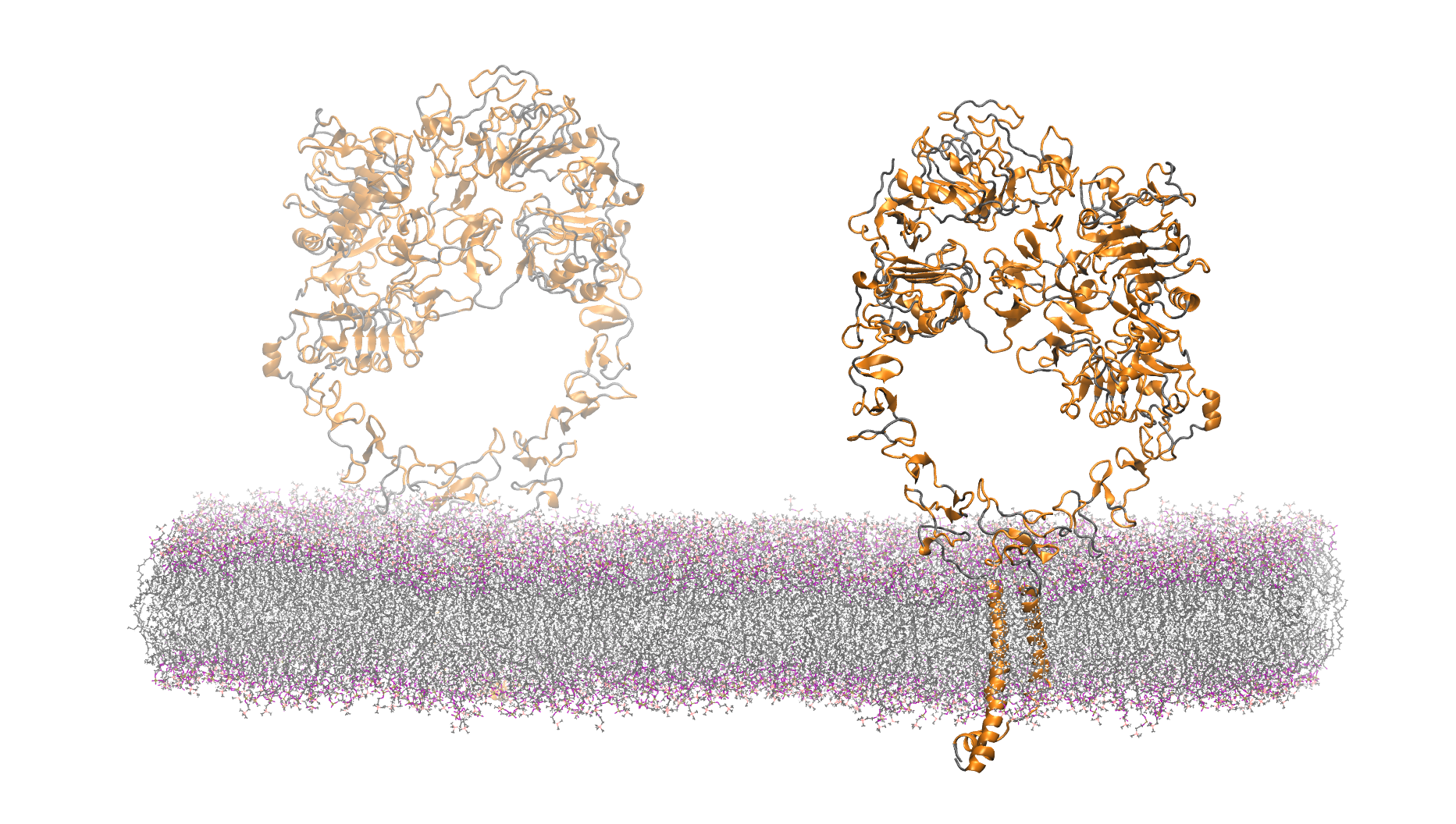
1.4M atom system - A Pair of hEGFR Dimers of 1IVO and 1NQL
Total number of atoms = 1,403,182
Protein atoms = 43,498 Lipid atoms = 235,304 Water atoms = 1,123,392 Ions = 986
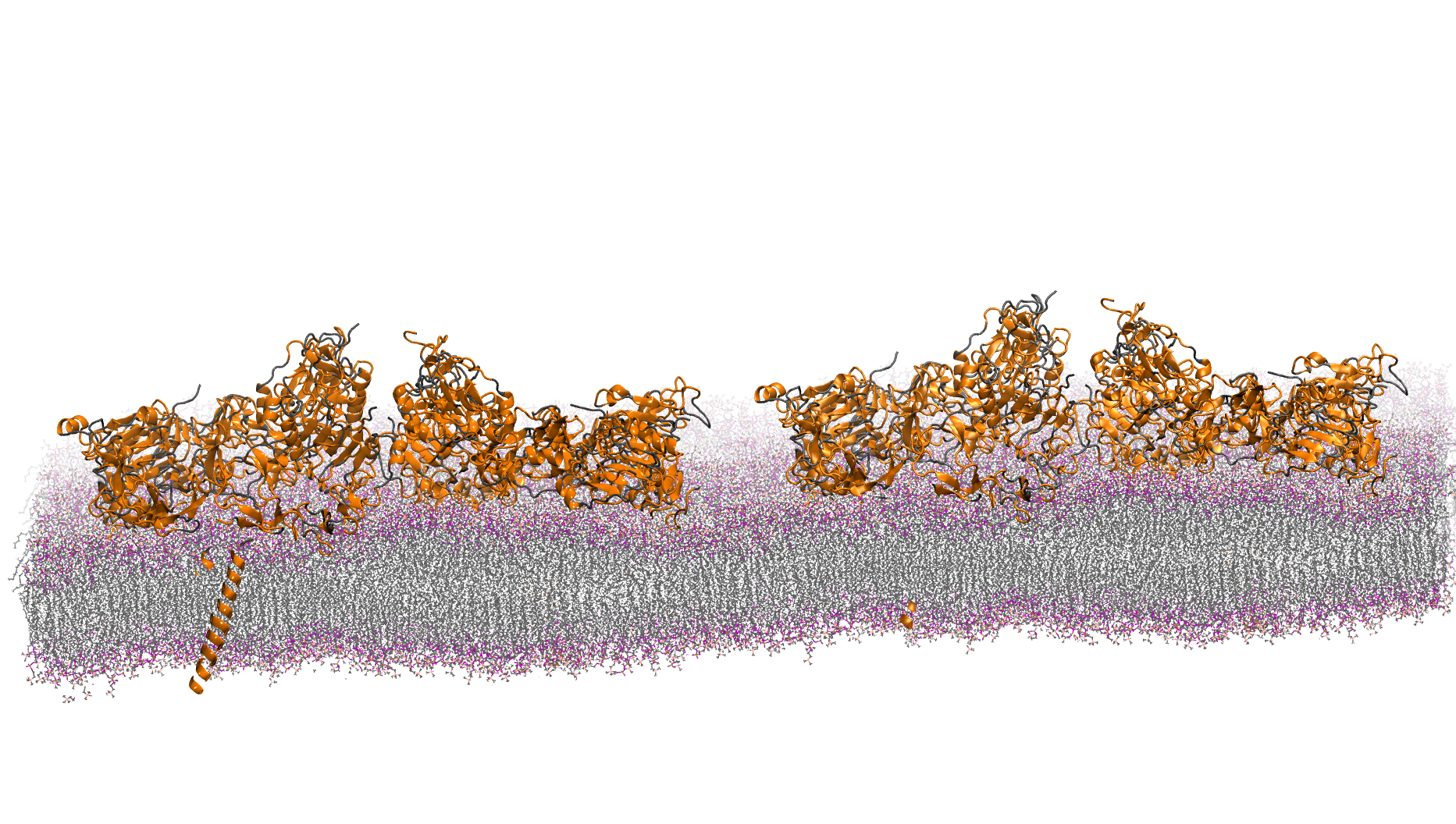
3M atom system - A Pair of hEGFR tetramers of 1IVO and 1NQL
Total number of atoms = 2,997,924
Protein atoms = 86,996 Lipid atoms = 867,784 Water atoms = 2,041,230 Ions = 1,914
The Benchmark Files
The following files make up our benchmarking suite, the files for each MD package are provided on an individual basis so that we can update for compatibiltity issues per each package release cycle.
AMBER - compatible with versions 16, 18 and 20
GROMACS - compatible with versions 5.1.x, 2016.x, 2018.x, 2019.x and 2020.x
LAMMPS - compatible with versions release in years 2016 through 2021
NAMD - compatible with versions 2.9 - 2.14 and 3.0-alpha