Contents
JADE2 (NVIDIA Tesla V100) Benchmarks
ARCHER2 (EPYC 7742) Benchmarks
Single node
Multiple nodes
OpenMP and MPI
Comparison across HPC systems
ARCHER2 and JADE2
JADE2 and GH200 testbed
Comparison of MD software across different HPC systems
Benchmark methodology
The benchmark input files taken from the HECBioSim benchmark suite. Benchmarks are run and plots are created using the hpcbench utility. The hpcbench repository also contains the SLURM input scripts used to run each simulation. Benchmarks were conducted using the version of each MD program available on each HPC system, so benchmarks running on different systems are not always running the same software version. More details on the method and more results can be found in the report 'Engineering Supercomputing Platforms for Biomolecular Applications'. Raw benchmark data can be found in the HECBioSim benchmark data repository (note: this data requires HPCbench to parse).
The plots in this section are given in terms of performance (ns/day) and energy usage (kWh/ns). The energy usage is calculated using the energy consumption data reported by SLURM.
Systems tested
| System (PDB codes) | No. of atoms | Protein atoms | Lipid atoms | Water atoms |
| 3NIR Crambin | 21k | 642 | 0 | 19k |
| 1WDN Glutamine-Binding Protein | 61k | 3.5k | 0 | 58k |
| hEGFR Dimer of 1IVO and 1NQL | 465k | 22k | 134k | 309k |
| Two hEGFR Dimers of 1IVO and 1NQL | 1.4M | 43k | 235k | 1.1M |
| Two hEGFR tetramers of 1IVO and 1NQL | 3M | 87k | 868k | 2M |
JADE2 (NVIDIA Tesla V100) Benchmarks
On JADE2, benchmarks were run on a single GPU. Though each JADE2 node contains eight NVIDIA Tesla V100 GPUs, the multi-GPU performance was poor. Benchmarks were run with a corresponding one-eigth of the node's available CPUs (dual Xeon e5-2698s). Performance of CPU-bound software (e.g. LAMMPS) suffers the most.


ARCHER2 Benchmarks
Single node
Each ARCHER2 node contains dual AMD EPYC 7742 CPUs for a total of 256 CPU cores. OpenMM was not tested on ARCHER2, as it is not designed to run on CPUs and only provides a reference CPU implementation.


Multi-node
Benchmarks were run on up to 16 ARCHER2 nodes. While the scaling is favourable (particularly for the larger systems), there is a significant cost to efficiency irrespective of the system and MD program.










MPI and OpenMP scaling
Different combinations of MPI and OpenMP were tested for GROMACS, NAMD and LAMMPS, which support mixed OpenMP and MPI parallelisation. For each figure, the legend shows how many OpenMP threads were used, with the rest of the parallelisation being handled with MPI processes. In almost all cases, pure MPI parallelisation was fastest.
GROMACS





NAMD





LAMMPS





GH200
The benchmark suite was run on the Nvidia Grace Hopper GH200 testbed at BEDE. Each GH200 chip contains a 'Hopper' GPU and a fast ARM Neoverse 96-core ARM CPU. Though the GH200 is marketed around AI, it provides excellent molecular dynamics performance and perfect compatibility with existing MD software.


LUMI-G (AMD MI250X)
OpenMM couldn't be tested on LUMI-G due to a temporary incompatibillity with the OpenMM ROCm plugin at time of testing. Other MD software ran on LUMI-G, though LAMMPS was only supported via Kokkos, which does not implement some fixes used in the benchmark. In general, performance on the MI250x is comparable to contemporary Nvidia GPUs, though power efficiency is worse.


Comparison between different HPC systems
ARCHER2 and JADE2


JADE2 and GH200 testbed


Comparison of MD software across different HPC systems








Memory usage


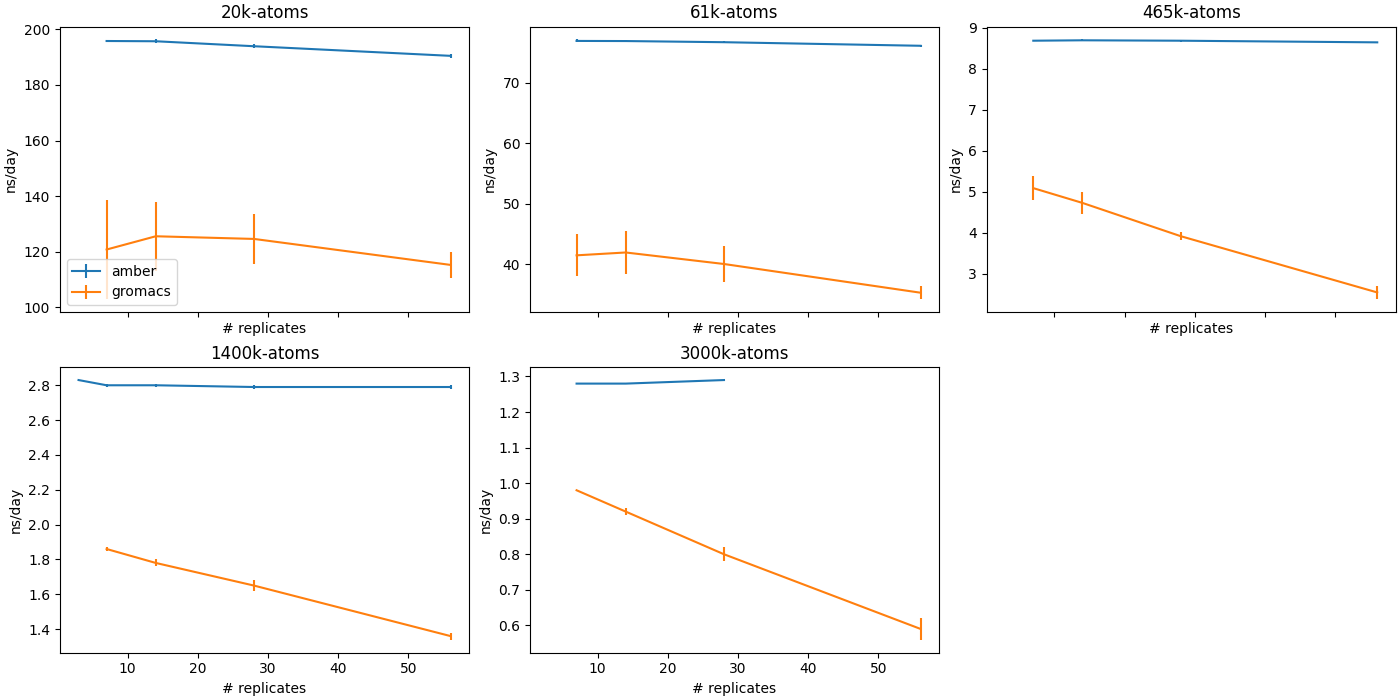
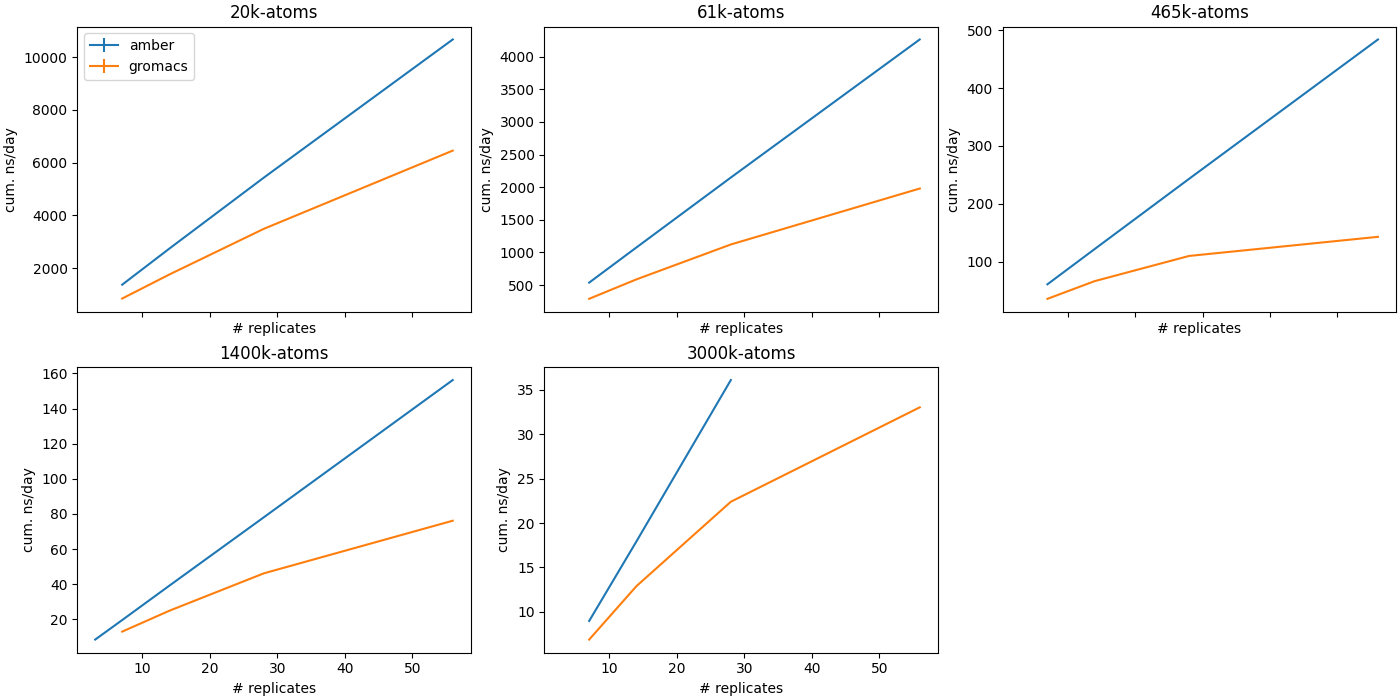
Multi-instance GPU (MIG)
MIG is an Nvidia GPU feature which allows single GPUs to be partitioned into multiple instances, which can be addressed as if they were separate GPUs. Particularly for smaller, less memory-intensive MD workloads, MIG can provide equivalent or slightly better cumulative ns/day than running a single exclusive MD job. GROMACS cannot scale to many replicas, though this is likely due to GROMACS being CPU-limited.