
COVID-19 has given biomolecular simulators a common cause. The sudden “normality” of on-line working has opened up new collaborations, because your office neighbour is no nearer than your colleague in a different country.
This has nucleated a number of new collaborative teams, unusually from the same discipline, working together and sharing their data, expertise and resources. Our alliance, seeded by the CCP/HECBioSim networks, combines the expertise of researchers spanning disciplines as broad as structural biology, medicinal chemistry, computational chemistry and theoretical physics, but all with a common set of biomolecular simulation and structural biology insights. We have found that independent teams working interactively towards a common goal can be extremely efficient in identifying and solving technical problems and ensuring reproducibility. Each research group has its own individual protocols and experience. Sharing these details with each other, and then comparing the results, has suddenly felt like the right way to do science. It provides confidence in the generality and reproducibility of our results, but also enables us to pool results to provide greater insight than any research team could do alone. Without exception, every contributor to the collaboration has identified a problem, and communally we have proposed and implemented solutions.
| The SARS CoV-2 viral helicase (nsp13): The viral helicase is the molecular machine responsible for separating the viral RNA from proteins and other RNA strands. Molecular dynamics simulations are being used to test the interaction of potential drugs, as well as to understand the mechanics of the motor, which may in turn lead to other drug design strategies. | |
 |
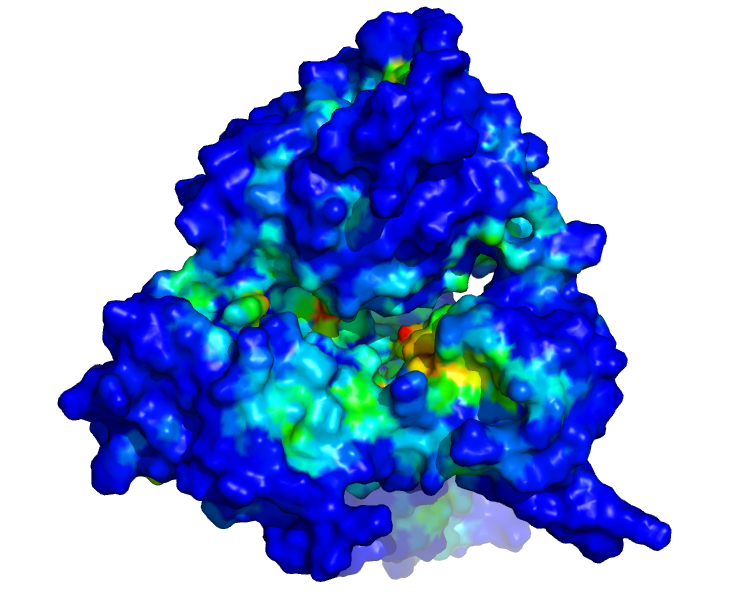 |
| Side view: Each colour shows a different molecular conformation, revealing the dynamic nature of the protein over microsecond timescales. | Top view: Colour coding illustrates the most druggable binding cavities, with red being the most favourable, and blue the least. |
Since the start of our alliance in March, we have been studying the viral helicase involved in COVID-19. Our groups have shared microseconds of simulation data and protocols used to generate them. We have developed expertise in the representation of metal ions in biomolecular structures, and we have used homology modeling to build biologically relevant complexes of the enzyme with its RNA and ATP substrates. We have shown that salt concentration in the simulations has a dramatic effect of the size and accessibility of binding pockets within the helicase, providing insight into the mechanistic importance of electrostatic interactions with the RNA and ATP substrates. We have used diverse and complementary methods for ligand docking and are currently pooling our results. We anticipate that our alliance will yield insights into the therapeutic potential of this target.
Our team:
Edina Rosta, Denes Berta, and Magd Badaoui: Computational chemists, King’s College London, UK
Elisa Frezza: Computational biophysicist from Université de Paris, CiTCoM, France, Paris
Nadia Elghobashi-Meinhardt: Computational biophysicist,Technische Universität Berlin, Germany
Andrei Pisliakov: Computational biophysicist, Physics and Computational Biology, Dundee, UK
Geoff Wells: Medicinal chemist from School of Pharmacy, UCL, UK
Sarah Harris: Computational biophysicist from the School of Physics and Astronomy and the Astbury Centre for Structural and Molecular Biology, Leeds, UK
Juliette Martin: Bioinformatician MMSB UMR 5086 CNRS/Université Lyon, France
Rachel Kerber: Computational chemist, Department of Chemistry, University of Cambridge, UK
Thomas Meltzer: Theoretical physicist, Department of Physics and Astronomy, UCL, UK.
Susanna Monti: Computational biophysicist, Istituto di Chimica dei Composti Organometallici, Pisa, Italy.
Christin Müller: Virologist, Justus Liebig University Giessen, Institute of Medical Virologi, Giessen, Germany
Mark Sanderson: X-ray crystallography, KCL, UK.
Andre Cobb: Synthetic chemistry, KCL, UK.
Arvind Ramanathan: Computational biologist, Argonne national laboratory/ consortium for advanced science and engineering (CASE), University of Chicago, USA.
The consortium would like to thank HecBioSim for time on the UK supercomputer ARCHER, AWS for cloud computing resources, and KCL for King's Together funding.